Mauro Pérez Cea, Miguel Peragón Cuesta, María Navarro Ojanguren.
1. Introducción
La secuenciación del ADN ha permitido estudiar la variación genética humana y su posible relación con enfermedades. El ADN es la molécula que almacena toda la información genética. Dentro del ADN hay regiones llamadas genes que contienen las instrucciones para fabricar una proteína o regular un proceso biológico. El ser humano tiene un total de 46 cromosomas, 23 pares, donde se almacena el ADN. Todos los cromosomas juntos forman un genoma. El genoma contiene todas las “instrucciones genéticas”, genes, regiones reguladoras… Lo que se va a usar en este blog, el exoma, es la parte del genoma formada por los exones, que son las regiones de los genes que sí codifican proteínas.
En este trabajo vamos a detectar las anomalías del exoma utilizando modelos generativos, concretamente Autoencoders Variacionales (VAE). Un VAE aprende a comprimir datos y luego reconstruirlos, comprimiendo cada dato en una distribución probabilística. Si el modelo aprende correctamente como son los perfiles genéticos esperados, aquellos perfiles que se construyan distinto serán considerados anómalos.
El objetivo principal del proyecto es construir un sistema capaz de identificar perfiles de variación genética atípicos en genes asociados a síndromes hereditarios de cáncer. Para ello se utilizan datos de referencia poblacional procedentes de gnomAD, una base de datos que recoge variantes genéticas observadas en muchos individuos. El dataset indica las frecuencias alélicas, que indican qué proporción de una población tiene una variante genética concreta. A partir de las frecuencias alélicas de estas variantes se simulan perfiles genéticos de referencia, que se utilizan para entrenar el VAE.
Posteriormente, el modelo se evalúa comparando estos perfiles esperados con perfiles que contienen una mayor carga de variantes raras o variantes clínicamente relevantes obtenidas de bases como ClinVar.
La idea no es realizar un diagnóstico médico, sino detectar perfiles genéticos anómalos en genes asociados a síndromes hereditarios de cáncer, tomando como referencia la variación poblacional de gnomAD.
2. Estado del arte
La interpretación de variantes genéticas es un problema central de la genómica actual. Bases de datos como gnomAD proporcionan frecuencias poblacionales que permiten estimar si una variante es común o rara, mientras que ClinVar aporta interpretación clínica de variantes concretas y sirve como fuente de validación. Los VAE han demostrado su utilidad en biomedicina para detección de anomalías y aprendizaje de estructuras latentes, siendo EVE el ejemplo más relevante: un modelo generativo no supervisado que aprende restricciones biológicas sin etiquetas clínicas. Este proyecto combina ambos enfoques, utilizando un VAE para aprender la distribución esperada de perfiles genómicos sanos y detectar desviaciones compatibles con síndromes hereditarios de cáncer
3. Desarrollo
El objetivo es detectar anomalías genómicas compatibles con tres síndromes hereditarios de cáncer: Lynch, HBOC y Li-Fraumeni, utilizando 12 genes representativos (MLH1, MSH2, MSH6, PMS2, EPCAM, BRCA1, BRCA2, PALB2, RAD51, CHEK2, TP53 y ATM). La arquitectura del pipeline es modular y escalable, permitiendo su extensión a cualquier conjunto de genes mediante la simple modificación de la lista de entrada.
Etapa 1: Delimitación de Ventanas Genómicas mediante Anotación (GTF)
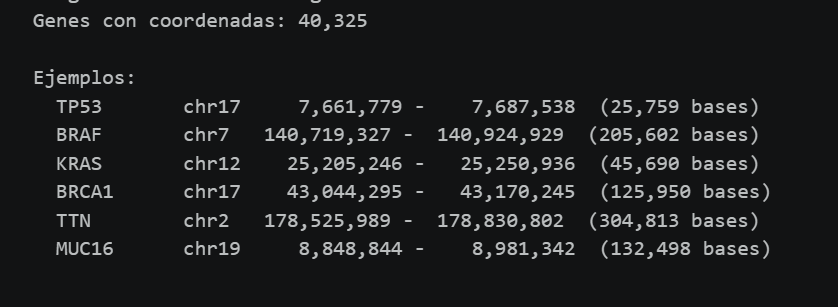
Antes de analizar cualquier variante genética, hay que conocer con precisión las coordenadas genómicas de cada gen. Para ello, utilizamos un archivo en formato GTF (Gene Transfer Format) de Ensembl. Con este archivo construimos una matriz de referencia con las coordenadas cromosómicas exactas (cromosoma, posición de inicio, posición de fin) de los genes.
Esta etapa permite al algoritmo definir “ventanas genómicas” específicas para cada gen, lo que posteriormente facilita la extracción de variantes mediante indexación tabix sobre los archivos VCF de gnomAD.
Etapa 2: Extracción y Agregación de Variantes por Gen
Una vez consolidada la matriz de coordenadas, el pipeline accede a los archivos VCF indexados de gnomAD (exomas) utilizando pysam y tabix. Para cada uno de los genes, el sistema recupera todas las variantes que caen dentro de sus coordenadas genómicas, verificando adicionalmente mediante el campo VEP (Variant Effect Predictor) que cada variante pertenece efectivamente al gen objetivo.
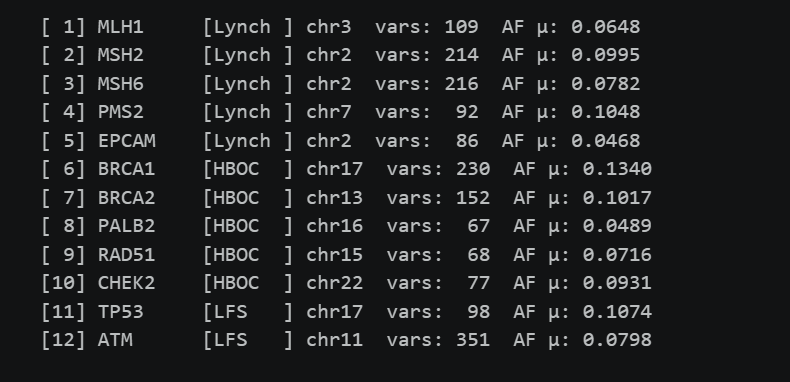
El valor diferencial está en la agregación funcional. En lugar de tratar el genoma como un conjunto de millones de mutaciones independientes, todas las variantes recuperadas dentro de un mismo gen se consolidan bajo una representación unificada. Cada gen queda caracterizado por tres métricas: el número de variantes raras (frecuencia alélica < 0.05), el número de variantes comunes (frecuencia alélica ≥ 0.05), y la frecuencia alélica media ponderada por el total de variantes en el gen.
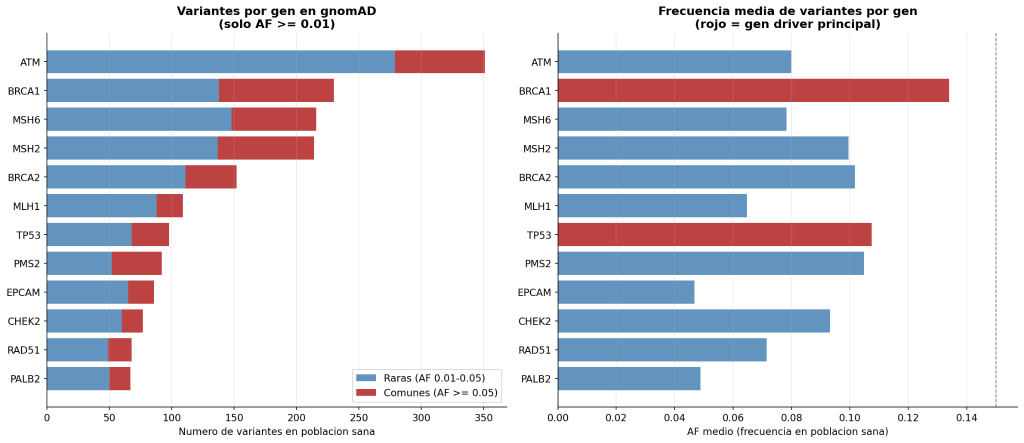
La Figura muestra el perfil de variantes de los 12 genes en población sana (gnomAD). Existe una heterogeneidad marcada entre genes: ATM y BRCA1 acumulan más de 250 variantes mientras que PALB2 y RAD51 apenas alcanzan 50, con frecuencias alélicas medias que oscilan entre 0.05 y 0.15 . Esta caracterización basal es la que el VAE aprende como perfil “normal” para detectar desviaciones patológicas durante la inferencia.
Etapa 3: Transformación Logarítmica y Normalización
Dado que las distribuciones de variantes presentan un sesgo natural extremo, los conteos se transforman logarítmicamente: log1p para variantes raras y comunes, y log10 para la frecuencia alélica media. Los vectores resultantes de dimensión 36 (12 genes × 3 features) se normalizan mediante StandardScaler (media cero, varianza unitaria) para garantizar un rango numérico estable en la entrada del VAE
Etapa 4: Entrenamiento del Autoencoder Variacional (VAE)
El VAE se entrena con 20.000 perfiles sintéticos generados mediante muestreo binomial sobre las frecuencias alélicas de gnomAD, aprendiendo la distribución esperada de la población sana sin sesgos médicos. La arquitectura consta de un encoder (36→72→36→16D) y un decoder simétrico, donde el espacio latente de 16 dimensiones captura las correlaciones entre los 12 genes estudiados
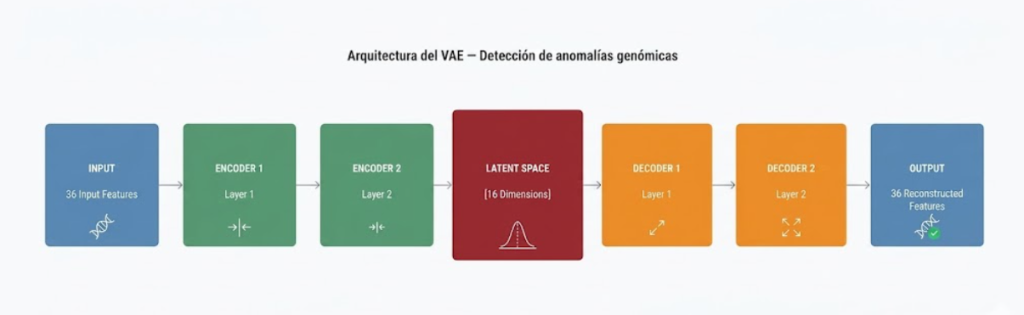
El entrenamiento utiliza la función de pérdida ELBO compuesta por error de reconstrucción MSE y divergencia KL ponderada por β=0.001, con Dropout de 0.2 y optimizador Adam durante un máximo de 200 épocas con early stopping tras 30 épocas sin mejora. El umbral de detección de anomalías se fija en el percentil 95 del error de reconstrucción sobre el conjunto de entrenamiento sano.
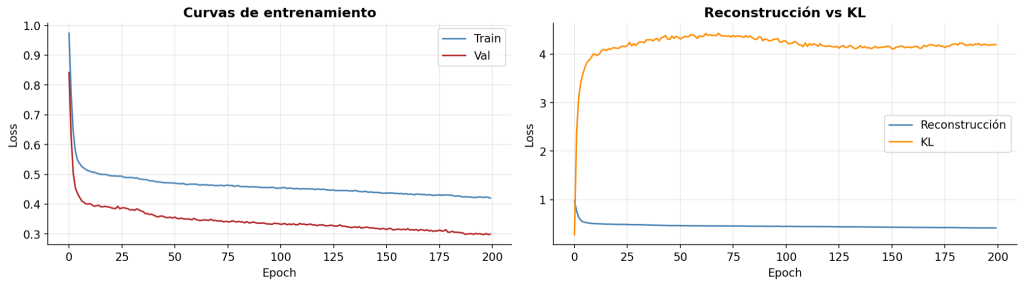
Las curvas de entrenamiento muestran una convergencia estable a lo largo de 200 épocas sin signos de overfitting, con la pérdida de validación estabilizándose en torno a 0.30. El error de reconstrucción MSE alcanza ~0.4 mientras que la divergencia KL, controlada por β=0.001, se estabiliza en ~4.2 sin colapsar el espacio latente, confirmando que las 16 dimensiones latentes codifican información biológica relevante.
Etapa 5: Inferencia y Generación de Reporte Clínico
Durante la inferencia, el perfil genómico del paciente se construye siguiendo el mismo pipeline de extracción, agregación y normalización, y el vector de 36 dimensiones resultante se introduce en el VAE. Si el error de reconstrucción MSE supera el percentil 95, el perfil se clasifica como anómalo y el sistema genera un reporte clínico que identifica los genes hipermutados, la intensidad de la alteración (leve: 1.2, moderada: 1.5, severa: 2.5) y el síndrome más probable por similitud coseno contra perfiles ClinVar de Lynch, HBOC y Li-Fraumeni.
4. Material
- Para este trabajo hemos utilizado cuatro fuentes de datos abiertas: gnomAD como referencia de variación genética poblacional, Ensembl para obtener las coordenadas cromosómicas de los genes estudiados, ClinVar para validación clínica de variantes patogénicas, y 1000 Genomes para validación con perfiles genéticos reales.
- Las herramientas de código empleadas son: pandas y numpy para manipulación de datos, matplotlib para visualización, pysam para lectura de archivos VCF, scikit-learn para normalización y métricas, PyTorch para implementar y entrenar el VAE, y UMAP para visualizar el espacio latente aprendido por el modelo.
5. Resultados
Para la evaluación hemos creado cuatro grupos de perfiles distintos: 200 sanos, generados con el mismo proceso binomial del entrenamiento y 400 anómalos distribuidos en tres niveles de dificultad de detección. Los perfiles leves (x1.2) simulan portadores con una carga de variantes raras muy poco superior a la de referencia de la población sana. Los moderados (x1.5) representan alteraciones moderadas, y los severos (x2.5) corresponden a perfiles con alta carga de variantes clínicamente significativas.
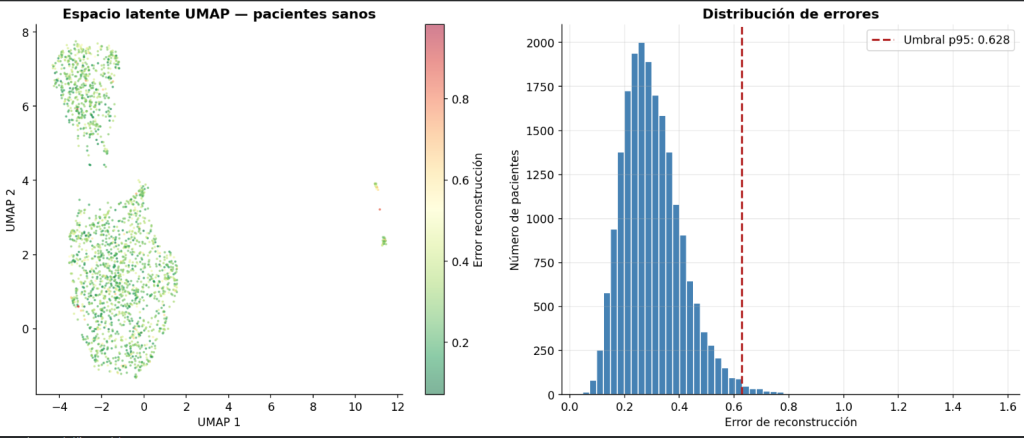
La clasificación se realiza de forma no supervisada: el único criterio es si el error de reconstrucción MSE supera o no el umbral del percentil 95 establecido durante el entrenamiento.
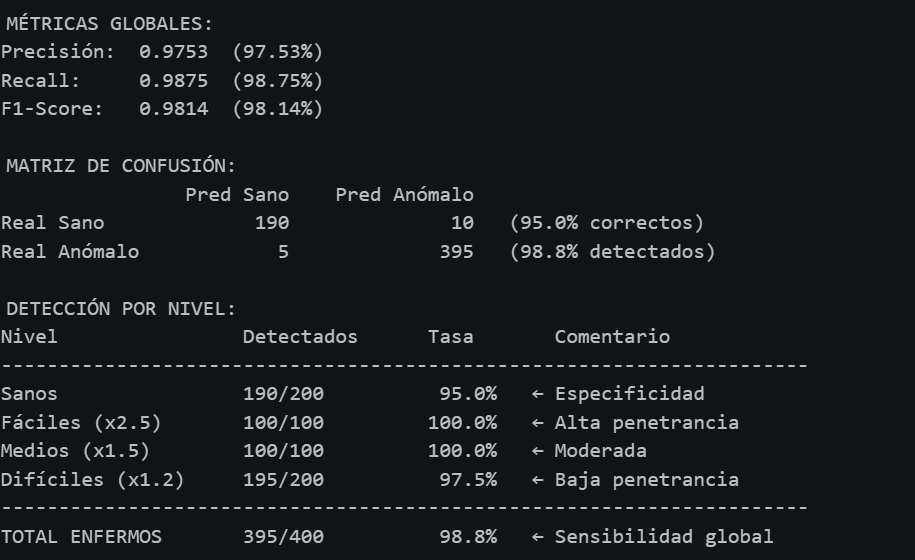
Los resultados globales muestran una precisión del 97.53%, un recall del 98.75% y un F1-Score del 98.14%. De los 200 perfiles sanos, 190 se clasificaron correctamente, lo que da una tasa del 95%, coherente con la definición que pusimos al umbral. De los 400 perfiles anómalos, el modelo detectó 395, con solo 5 falsos negativos, todos ellos de la categoría más difícil (x1.2).
Para la visualización del espacio latente evaluamos 15 pacientes independientes: 5 perfiles sanos generados con semilla independiente no utilizada durante el entrenamiento, 5 pacientes Lynch y de 5 HBOC con 8 variantes reales extraídas de ClinVar.
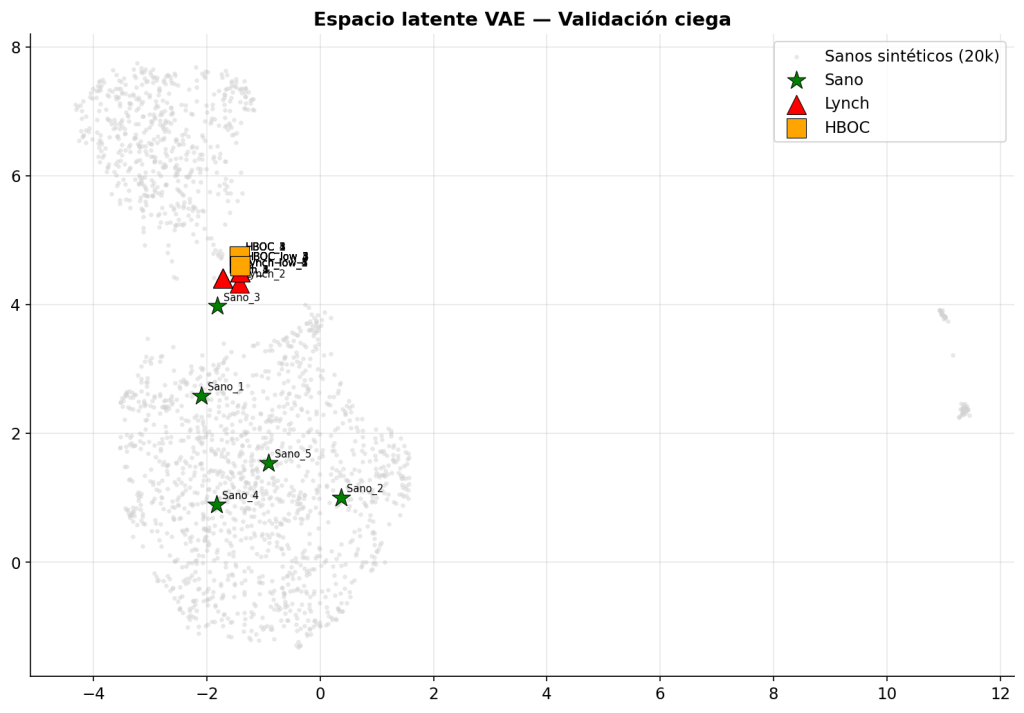
Los resultados fueron que los sanos se integran en la nube de puntos de los 20000 sanos sintéticos mientras que los pacientes con genes alterados de Lynch y HBOC se proyectaron en una región distinta.
6. Discusión de resultados
Los resultados demuestran que un VAE entrenado exclusivamente sobre perfiles genómicos de población sana es capaz de detectar anomalías asociadas a diferentes enfermedades sin haber visto ningún ejemplo de esas enfermedades durante el entrenamiento. La capacidad de detección se debe a haber aprendido qué distribución de variantes es normal, no de memorizar patrones de enfermedad. Que el sistema detecte correctamente casi todos los perfiles anómalos (97.5%), sugiere que el VAE ha aprendido a reconocer el patrón global de un genoma normal.
Sin embargo, el trabajo tiene algunas limitaciones prácticas. Tanto los datos de entrenamiento como los de evaluación son sintéticos, generados a partir de frecuencias poblacionales de gnomAD. En datos reales de pacientes, existen diferentes factores que podrían introducir ruido al modelo, que bajaría la precisión del modelo.
El punto más importante es que la anomalía detectada no equivale a una enfermedad concreta (solo para el ejemplo de Lynch y HBOC añadida como imagen en el espacio latente). El sistema identifica perfiles inusuales respecto a la referencia poblacional, no enfermedades. El valor que aporta el trabajo consiste en identificar perfiles fuera de lo común y se consideran como “perfiles de riesgo” que merecen atención.
7. Referencias
- Genome Aggregation Database (gnomAD). gnomAD v4.0 / v4.1 release information. (gnomad.broadinstitute.org)
- Landrum, M. J., Lee, J. M., Benson, M., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067.
(CNBiotecnología) - Frazer, J., Notin, P., Dias, M., et al. (2021). Disease variant prediction with deep generative models of evolutionary data. Nature, 599, 91–95. (PubMed)
- The 1000 Genomes Project Consortium. (2015). A global reference for human genetic variation. Nature, 526, 68–74. (Nature)
- Kingma, D. P., & Welling, M. (2013). Auto-Encoding Variational Bayes. arXiv:1312.6114.
- Ensembl Genome Browser. Human genome annotation GRCh38.